Protein Folding
Introduction
This Python script is dedicated to a research project during the summer. The goal of this project is to find the best way to fold proteins using a hydrophobic-polar protein folding model. The concept of the “best way” refers to a method for transforming a string into 2D or 3D shapes that best satisfy the criteria of hydrophobic-polar protein folding.
hydrophobic-polar protein folding model
The hydrophobic-polar protein folding model is a highly simplified model for examining protein folds in space, first proposed by Ken Dill in 1985. In this model, a protein is represented as a string containing 1s and 0s. Between each 1 or 0, there is a connection point where the protein can be folded.
In order to score, the following condition must be satisfied:
- The number must be next to each other without a connection
- Both number must be 0
To Better illustrate this idea, Let’s take a look at a example
example
Given the input is [0, 0, 0, 0, 0, 0, 0, 0], the script will produce:
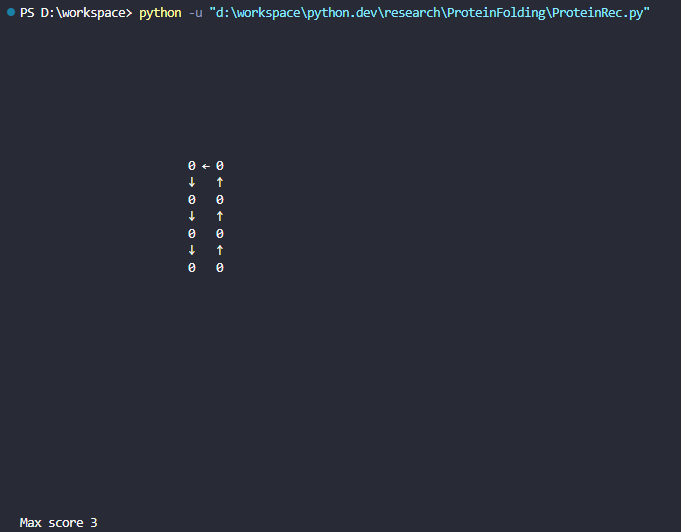
One of the best ways to fold the protein is in the image. As you can see, this 2D shape scores 3 because there are 3 pairs of 0 next to each other without connection (arrow).
method
To achieve this goal, we utilize recursive backtracking. First, we choose the very first number in the input and place it in an arbitrary position on the grid. Then, we use a for loop to check the positions around the first number. We place the second number in the first valid position we find, and we continue this process. When the script encounters a situation where there are no valid positions, it will recurse and move to the next valid position. The script will terminate when all the numbers have been placed on the grid or when there is no available space for any more numbers.
Code
Before I introduce the main part of the script, I would like to first introduce some helper fuction.
isValid()
This function simply checks if the block on the grid is occupied or not.
def isValid(grid, y, x):
if grid[y][x] == " ":
return True
return False
Checkpts()
As we are plotting the 1s and 0s, this function checks if there are any scores around the current 1s or 0s that we are plotting.
def Checkpts(grid, y, x, direc):
if direc == "R":
try:
if (grid[y][x] == 0 and grid[y][x + 2] == 0):
return 1
elif (grid[y][x] == 0 and grid[y - 2][x] == 0):
return 1
if (grid[y][x] == 0 and grid[y + 2][x] == 0):
return 1
else:
return 0
except:
return 0
elif direc == "L":
try:
if (grid[y][x] == 0 and grid[y][x - 2] == 0):
return 1
elif (grid[y][x] == 0 and grid[y - 2][x] == 0):
return 1
elif (grid[y][x] == 0 and grid[y + 2][x] == 0):
return 1
else:
return 0
except:
return 0
elif direc == "U":
try:
if (grid[y][x] == 0 and grid[y - 2][x] == 0):
return 1
elif (grid[y][x] == 0 and grid[y][x - 2] == 0):
return 1
elif (grid[y][x] == 0 and grid[y][x + 2] == 0):
return 1
else:
return 0
except:
return 0
elif direc == "D":
try:
if (grid[y][x] == 0 and grid[y + 2][x] == 0):
return 1
elif (grid[y][x] == 0 and grid[y][x - 2] == 0):
return 1
elif (grid[y][x] == 0 and grid[y][x + 2] == 0):
return 1
else:
return 0
except:
return 0
ProteinFolding()
The ProteinFolding is the main function that will plot 0s and 1s on grid and check the score by using recursive backtracking. To explain this function clarly, I will break the function into small portions.
set up and base case
This function will take a couple parameters.
- Acidseq: The protein sequence from input.
- Grid: The temporary grid that stores all the 0s and 1s.
- Curnum: The current number that the script is plotting.
- Y: Vertical position axis of the grid.
- X: Horizontal position axis of the grid.
- Pts: How many points the script scores.
- Resgrid: The result grid.
The function’s base case is when the script reach the last 0 or 1 in the sequence. Once it reach it, the function will end.
def ProteinFolding(Acidseq, grid, Curnum, y, x, pts, resgrid):
global Maxpts
global y_move
global x_move
global direc
if Curnum == len(Acidseq):
if pts > Maxpts:
Maxpts = pts
CopyArray(grid, resgrid, len(grid))
return resgrid
plotting
Since the script is folding protein on a 2D grid, for each 1 or 0, there are 4 postion that the script can plot on. (left, right up, or down). To attempt each position, there is a for loop to iterate the portion.
for i in range(4):
y_prog = y + y_move[i]
x_prog = x + x_move[i]
y_new = y_prog + y_move[i]
x_new = x_prog + x_move[i]
After we figure out which position we want to attempt, the script will first check if that is a valid position to plot. If it’s not valid, this iteration will end and move on to the next one. If the position is valid, the script will place the current number at the position and check if there are many possible points around it. Finally, the script will call the function again but with the next number.
if isValid(grid, y_new, x_new):
grid[y_prog][x_prog] = symbol[i]
grid[y_new][x_new] = Acidseq[Curnum]
pts_new = pts + Checkpts(grid, y_new, x_new, direc[i])
CopyArray(resgrid, ProteinFolding(Acidseq, grid, Curnum + 1, y_new, x_new, pts_new, resgrid), len(grid))
grid[y_new][x_new] = " "
grid[y_prog][x_prog] = " "
return resgrid
Wrap up
After the script attempts all the possibilities, it will print out one of the best folds that it found. One drawback of this script is that it requires a significantly long time to explore all possibilities. As the input string gets longer and longer, the script will eventually take too much time to produce a result. To address this problem, we can adopt a more efficient algorithm or employ AI to reduce the time spent.
Source: Protein Folding